11/08/2020
Covid-19 sparks medical device boom – sheds light on device requirements

The Covid-19 pandemic has brought the whole world to a practical standstill, while healthcare is operating just beyond its limits. Currently, there are not enough health care staff to treat all Covid-19 patients as well as a shortage of much needed medical devices. This presents vast opportunities for established players and new entrants. Proper protocols need to be followed in product development, however, to ensure that devices are safe and reliable.
Covid-19 is caused by a coronavirus (SARS-CoV-2) and results in problems virtually in the whole body, the most serious of which occur in the lungs. A wide variety of devices can be used to ensure the functioning of the respiratory system of a Covid-19 patient. Unfortunately, there are too many Covid-19 patients, which has greatly fuelled demand for medical devices and created an unprecedented need for new innovations.
Medical device development a demanding process
Getting a medical device to market may seem cumbersome, time consuming and a little daunting. The process is indeed demanding, but it is intended to ensure that products launched on the market have been properly designed, are safe and reliable. There is a natural temptation to look for shortcuts or alternatives, but this should be avoided. In most cases, such approaches are doomed to cause delays and increase costs further down the road.
High demands are a requirement even now during the pandemic, although the need for fast equipment availability has grown tremendously. The path to the market may be faster at the moment, but the associated quality standards remain high.
Consider the following scenario: Company A has an excellent product idea, a product design or even a product prototype. The market is also ready for the product and a clear need exists.
When considering how to develop and get the product launched, Company A begins to consider whether a medical device classification for the product will result in an unnecessarily expensive and lengthy project. What if competitors use this time to fill the space? Would it not be better to avoid the medical device classification and fast-track development, thereby ensuring success? In short, the answer is a resounding NO.
If a device is erroneously classified as serving a purpose other than that of a medical device, and the classification needs to be changed later, the entire process needs to be started from scratch. It makes much more sense to reduce uncertainty by adhering to proper processes.
There are seven phases involved in developing and launching a medical device successfully, starting from defining the device use and product classification, as well as its essential requirements, and ending with registering the device and product to market. A detailed overview of the phases follows overleaf.
Phase 1: Defining device use and product classification
In the first phase of getting a medical product to market, the device designer or manufacturer must define the device use. In practice, this is where the manufacturer decides whether the device is a medical device or not.
The definition of use also defines the entire process of getting the product to market and, therefore, this phase requires special attention. In Europe, guidelines for defining use are provided in the European Medical Devices Directive 93/42/EEC.
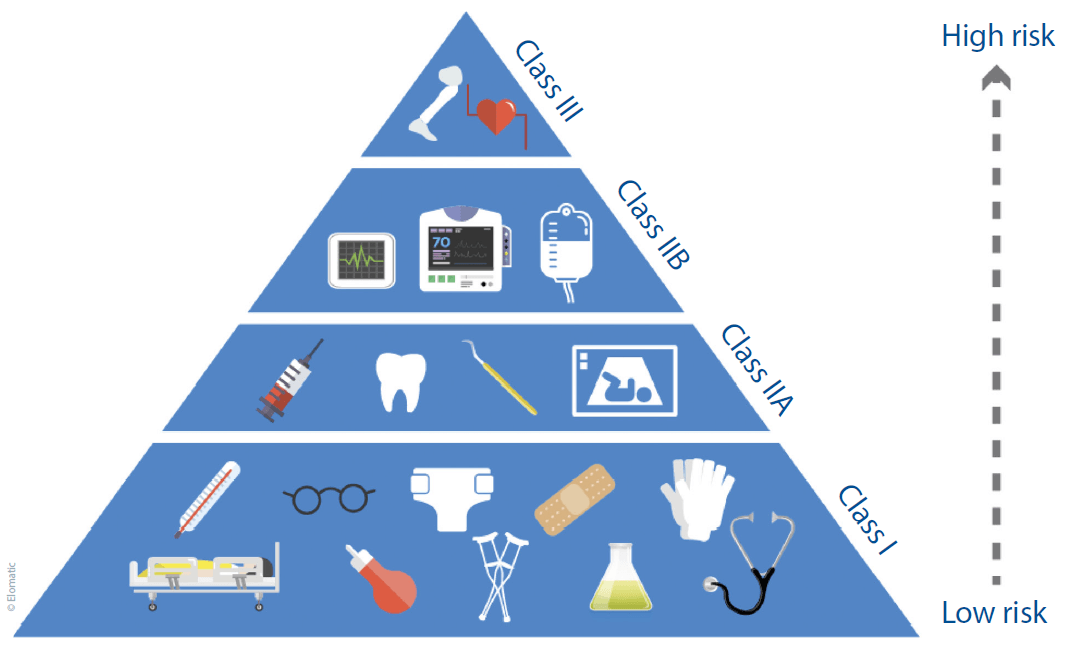
Once a device has been defined as a medical device, the manufacturer is obliged to classify the device appropriately according to the directive (see Figure 1). The EU’s Medical Devices Info internet pages (http://meddev.info/) are an invaluable resource in this regard, and manufacturers and developers are well advised to make full use of it.
Phase 2: Fulfilling essential requirements of device
The manufacturer is responsible for ensuring that the medical device fulfils the essential requirements as set out in the Medical Devices Directive for its intended use. The directive defines which standards are to be adhered to when designing, manufacturing and storing the device. The European commission also maintains and updates these harmonised standards on its web pages.
By fulfilling the essential requirements, the manufacturer ensures that the device can meet its defined performance levels and does not endanger the lives of patients and users. For this reason, the manufacturer also must perform a risk analysis of patient and user safety for the device.
Phase 3: Clinical evaluation
The manufacturer must specify the intended use of the product. The product description must also provide information about the characteristics and performance of the product in its normal environment of use. The demonstration of product compliance must include a clinical evaluation, which includes both an evaluation of the efficacy and functionality of the device. Any adverse effects should be identified and evaluated based on a risk analysis. A clinical evaluation is mandatory for all products.
A clinical evaluation determines, if necessary, the functionality and suitability for use of the device and its accessories. It determines and evaluates the properties, performance, and side effects of the device and its accessories. It is part of the procedure of attesting conformity before it is placed on the market or put into service.
The requirements for clinical evaluation are included in national legislation. The SFS-EN ISO 14155 standard can be used to assist in the design and implementation of research.
Phase 4: User and safety instructions
The device or its packaging needs to be labelled with information regarding the manufacturer and safe use. High quality user instructions should also accompany the product. The level of required detail in labelling usually becomes clear when fulfilling the essential requirements of the directive in phase 2.
In Finland, instructions for safe use intended for end users and/or patients are required in both Finnish and Swedish. General user instructions or similar information is required in Finnish, which is to be supplemented with either Swedish or English.
Legislation requires that high quality user instructions are drawn up and provided with medical devices. The proper use and operation of medical devices is essential from the perspective of the patient’s and user’s safety. It is, therefore, imperative that enough time and effort is put into drawing up such instructions.
If necessary, the help of experts with experience in producing high quality instructions should be considered. The quality of instructions is one of the areas the authorities consider when evaluating whether a device complies with the set requirements.
Phase 5: Evaluation of compliance with requirements
The manufacturer is obliged to draw up technical documentation regarding the fulfilment of essential requirements, as well as about the measures that enable the evaluation of the device’s compliance with requirements.
A clinical trial should always be conducted in order to verify compliance with requirements. The clinical approximate values define the device features and performance. The results of the risk analysis in phase 2 are also evaluated during the clinical device trial.
Specific procedures have been set out for different device classifications to prove compliance with the requirements. The provisions for acceptable clinical device trials are set out in national legislation. It is mandatory for all medical devices to undergo clinical trials.
For devices with IIA, IIB and III classifications the evaluation of compliance with requirements can be conducted only by facilities that have been declared by EU country governments. In Finland, there are currently two such facilities.
Phase 6: CE marking
When the compliance with requirements has been evaluated and proven, the manufacturer can attach a CE marking to the device. By attaching the marking the manufacturer confirms that the device fulfils its defined essential requirements. The marking also confirms that the manufacturer accepts responsibility for the design and manufacture of the product.
Phase 7: Registering device and product to market
When the first five phases have been completed, the manufacturer must register the device with the appropriate national authorities. In Finland, Fimea (https://www.fimea.fi) is responsible for maintaining the device register.
At this stage, one can say that the manufacturer has diligently followed the process and taken responsibility for ensuring that the device meets its defined requirements. It also at this stage the product could be considered ready for the market.
The marketing and launching of the medical device is in itself a challenging project, but if the process described in this article has been followed, the manufacturer can at least feel that all bases have been covered before the actual marketing and launch of the product.
Want to know more? Check out these related articles:
Pharma
We offer strong expertise in pharmaceutical and medical device production suites.
We help you in designing, constructing, and qualifying new facilities and production lines. Our services encompass all investment phases, from creating a concept to delivering the final report.
Compliance
The manufacturing of pharmaceuticals is highly regulated. We help you navigate through the requirements and tailor facilities according to your needs.